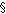
Zhaolei Zhangab, Lishar Huangab, Young-In Chib, Kyeong Kyu Kimb, Li-Wei Hungb, Antony R. Croftsc, Edward A Berrya, and Sung-Hou Kimab
Lawrence Berkeley National Laboratorya,
Department of Chemistry, University of California at Berkeleyb, and
Center for Biophysics and Computational Biology, University of Illinois at Urbana-Champaignc
This paper is dedicated to the memory of our friend and
colleague Vladimir M. Shulmeister, who was the principle crystallographer on
the project until his untimely death on September 27 1995.
Xray crystallographic analysis of multiple crystal forms of the cytochrome bc1 complex from three different species reveals two different locations for the ironsulfur protein (ISP). One location is close enough to the putative quinol oxidation site to allow reduction of the ISP by ubiquinol. The other site is close enough to cytochrome c1 to allow rapid oxidation of the ISP by the cytochrome. However neither location would allow both reactions to proceed at a kinetically competent rate. The reaction mechanism thus must involve a dramatic conformational change involving a large movement of the ironsulfur protein.
A basic principle of aerobic respiration in eukaryotes and bacteria, as well as oxygenic or cyclic photosynthesis, is the coupling of electron transfer along a chain of redox components to proton translocation across the membrane in which those redox components are embedded. This gives rise to an electrochemical proton gradient across that membrane, which can be coupled to ATP synthesis and other energy-requiring processes. This principle was first proposed by Peter Mitchell in his chemiosmotic hypothesis (1). The mammalian mitochondrial respiratory chain was fractionated into four membrane protein complexes carrying out different segments of the electron transfer pathway (2). These were designated complexes I, II, III, and IV.
In mitochondria and in many aerobic or photosynthetic bacteria the central component of the electron transfer chain is a complex of membrane proteins known as the cytochrome bc1 complex, or ubiquinol:cytochrome c oxidoreductase (E.C. 1.10.2.2), equivalent to complex III in the mitochondrial respiratory chain. This enzyme catalyzes electron transport from ubiquinol to cytochrome c coupled to translocation of 2 H+ across the inner mitochondrial membrane, and the release of 2 additional H+ to the inter-membrane space, per quinol oxidized (3, 4, 5). The beef heart complex consists of 11 polypeptides (6, 7) with a total molecular mass of 240 kDa. There are four redox centers: two b cytochromes, cytochrome c1, and the Rieske iron-sulfur cluster.
A mechanism accounting quantitatively for the proton translocation coupled to electron transport by this enzyme and consistent with a number of otherwise difficult-to-explain experimental observations is a version of the "protonmotive Q cycle" of Peter Mitchell (4, 5).
Until recently only a low resolution structure for the complex from Neurospora crassa has been available from electron microscopy of 2-dimensional crystals (8). In 1991 Yu's laboratory reported 3-D crystallization of cytochrome reductase from beef mitochondria in a tetragonal space group (9). Two other groups independently reported crystallization of the complex in different space groups in the following year (10, 11). Recently Xia et al. reported a partial structure of the complex based on the tetragonal crystals (12, 13). However these crystals were somewhat disordered in the extrinsic intermembrane domains, including the functional domains of cytochrome c1 and the Rieske ironsulfur protein. Thus while the Fe2S2 cluster was located by anomalous scattering, no model was presented for the extrinsic domain of the ironsulfur protein(13) despite availability of the coordinates of this domain from crystals of a soluble fragment(14).
In the meantime we have developed a new monoclinic crystal form of the beef bc1 complex (15) and crystallized the complex from other vertebrate sources (Table 1). The rabbit complex gave crystals in the same hexagonal space group as the hexagonal beef crystals, but with significantly different cell parameters and with slightly better order, diffracting to 3.5 A. The chicken complex crystals were in the orthorhombic space group P212121 and diffracted to 3.0 Å. We have found conditions for freezing the chicken crystals without significant loss in resolution, for cryogenic data collection.
The chicken crystal data were phased by isomorphous replacement and the resulting electron density was used to phase three other crystal forms by molecular replacement. The phases were improved by multi-crystal and noncrystallographic symmetry averaging until an atomic model of the protein could be built into the electron density. The apparent membrane-spanning region contains 12 transmembrane helices. It was possible to locate the structure (14) of the Rieske iron-sulfur protein (disordered in the structure of ref. s13 from beef tetragonal crystals) in the density. Atomic models were built de novo for all the other subunits except subunit 9 and subunit 11. In the density maps from crystals of beef or rabbit protein a 13th transmembrane helix is present. We attribute this to subunit 11 which seems to be absent from our chicken preparation.
We were able to trace cytochrome c1 to obtain its folding pattern for the first time. The core structure resembles that of Ambler's type I cytochromes c, such as miotochondrial cytochrome c or bacterial cytochrome c2, with insertions and deletions in peripheral loops accounting for the difference in size. Deletion of the loop corresponding to residues 41 to 58 of tuna cytochrome c exposes the heme propionates and opens a second pathway for electron transfer by cytochrome c1. We believe this pathway is used for reduction of cytochrome c1 by the Rieske ironsulfur protein.
The position of the ironsulfur protein is slightly different in the different crystal forms of the native protein. In both forms of our beef crystals, the distance between the cytochrome c1 heme and the ligand field of the ironsulfur cluster is small enough to allow rapid electron transfer between cytochrome c1 and the ironsulfur protein in this conformation, as experimentally observed(5, 16, 17). However the distance from the Rieske cluster to the presumed ubiquinol binding site is too great to allow rapid electron transfer.
When the Qo-site inhibitor stigmatellin is bound to the chicken complex before crystallization, the ironsulfur protein moves, putting the ironsulfur cluster closer to cytochrome b, near the position found in the tetragonal beef crystals of Xia et al. (13). This conformation would be expected to allow rapid electron transfer from ubiquinol to the ironsulfur protein, perhaps via a hydrogen bond from ubiquinol to a cluster-ligating histidine as proposed (18, s19, s20).
Other Qo-site inhibitors (myxothiazol, MOA-stilbene) bind in a different, but overlapping site in the pocket, closer to the low-potential heme of cytochrome b, allowing electron transfer from ubiquinol or ubisemiquinone at this position to the heme. Density due to bound antimycin is adjacent to the high potential heme of cytochrome b. In the presence of these inhibitors the ironsulfur protein is in the same position as in the native crystals.
Taken together our two structures for the ironsulfur protein and 3 positions for ubiquinone analogs are compatible with all the reactions proposed by the Q-cycle mechanism for electron transfer coupled to proton translocation, however no one structure alone would be competent. We therefore propose that the reaction mechanism requires a dramatic conformational change involving movement of the ironsulfur protein.
RESULTS:
Map quality and interpretation, overall shape of the cytochrome bc1 dimer-
During the phase improvement and extension process (see methods), correlation coefficients between the calculated map of the Rieske protein and our electron density increased to 80-85% in the different crystals, while the correlation coefficient between subunits 1 and 2 increased to 40-48% for different crystal datasets. Figure 1 shows density in the region of the heme of cytochrome c1, contoured at 1.5 sigma, at different stages during phase improvement and extension by averaging. In maps from the final improved and extended experimental phases (Fig. 1, c and d), the propionate side chains and thioether linkages are clearly visible.
Figure 2 shows our current model of the bc1 complex dimer in three different orientations. The monomers are related by a 2-fold axis running vertically in the plane of the paper. This same relation between monomers in the dimer was observed in all crystal forms, although in some cases the dimer 2-fold axis fell on a crystallographic 2-fold resulting in a monomer in the asymmetric unit. Our detergent-solubilized complex has a molecular weight around 500 (estimated by gel filtration), and we presume the same dimeric relation observed in the crystals reflects the situation in the mitochondrial membrane.
The overall shape is the same as described for the beef complex (12, 13), but considerably more protein is present in the intermembrane domain. We have located subunits 1 through 8 and 10 in the density of the chicken crystal. As discussed below, subunit 11 seems not to be present in our preparation. We also see density which we attribute to ubiquinone and phospholipid at a number of sites in the transmembrane portion. The presumed transmembrane segment (see below) is indicated by the band of gray representing a lipid membrane. The protein extends from the membrane 79 Å into the matrix space and 31 Å into the inter-membrane region on either side of a transmembrane region 40 Å thick, giving a total length of 150 Å perpendicular to the membrane.
Assignment of redox centers and membrane sidedness:
A Bivoet difference map made from data collected at 7131 eV (near the iron K absorption edge) revealed 8 iron centers in the asymmetric unit, as expected for a dimer of a protein containing three hemes and one Fe2S2 cluster.
A region composed mainly of roughly parallel alpha helices was assumed to be the transmembrane region. Four iron centers (from the anomalous Bivoet difference map) enclosed within two 4-helix bundles in this region were interpreted to be the hemes of two cytochromes b in the dimer. The other iron sites were on one side of the transmembrane region. This side was assigned to be the outside, i.e. intermembrane side, based on biochemical evidence indicating that cytochrome c1 and the Rieske ironsulfur center are located on the external surface of the inner mitochondrial membrane (e.g. ref. 21). As phases were improved and extended one of these took on the shape of a heme (Figure 1) and the protein around the other could be correlated with the known structure of the Rieske ironsulfur protein, as described below.
Assignment of transmembrane helices.
Twelve transmembrane helices were found in the chicken complex. Their positions at two levels in the membrane are shown in Figure 3a and b,. Eight of the transmembrane helices were assigned to cytochrome b. The other 4 belong to cytochrome c1, the Rieske ironsulfur protein, and subunits 7 and 10; each of which has one transmembrane helix.
The secondary structure and folding pattern of cytochrome b is schematized in Figure 3d. The presence of 8 transmembrane helices in cytochrome b had been predicted based on sequence analysis(22-s24). We have adopted the nomenclature used by these authors: capital letters A-H for the transmembrane helices, and lower case letters (a, cd, ef etc.) for the linker regions between transmembrane helices. The amphipathic cd helix turned out to be two helices in a hairpin arrangement, designated cd1 and cd2 (13). It is customary in aligning sequences of cytochrome b to use the numbering of the Sacharomyces cerevisiae protein. Given the number of a residue in the chicken sequence (used here), the number of the corresponding residue in the yeast sequence is found by subtracting 2 if the number is less than 114. For residue 114 and later the numbering is the same as in yeast. The number of the corresponding residue in the beef sequence is found by subtracting 1 from the number in the chicken sequence after the first 5 residues. These algorithms are based on the alignments of reference 24.
Transmembrane helixes A-D of cytochrome b form a 4-helix bundle which encloses and provides the axial ligands for the hemes of cytochrome b, indicated in figure 3a and 3b. The binding sites of the inhibitors antimycin and stigmatellin are also indicated.
The positions and assignment of the transmembrane helices agree with those reported for the beef structure (13) except for our subunit 10 which corresponds to their unassigned helix 1; and their unassigned helix 2 which is absent in the electron density from our chicken crystals. Examination of this region in the three mammalian crystals indicates the presence of a thirteenth transmembrane helix in the position of unassigned helix 2 of the beef structure(13). It probably corresponds to subunit 11, which we have been unable to identify by gel electrophoresis or reverse-phase HPLC(25) in our chicken preparation. Subunit 9, which is present in the preparation, has not been located in the density, and is also missing from the structure of reference R10. This subunit is the pre-sequence (26) of the Rieske protein, which gets cleaved off by a matrix processing protease, so it is likely to be on the matrix side of the membrane.
In the transmembrane domain the helices of the dimer fall into two clearly separated bundles. While the assignment of 2-fold related subunits to monomers is in principle arbitrary, it seems reasonable to divide the dimer along a line passing vertically through the center of symmetry in Figures 3a and 3b so that one monomer corresponds to one bundle of helices. We assign the chain letters A through K to subunits 1 through 11 of one monomer (the one whose cytochrome b heme iron coordinates have the smaller value in the x-coordinate), and N through X to subunits 1-11 of the other monomer. As described above subunit 9 (chains I and V) has not been located yet, and subunit 11 (K and X) is tentatively assigned to a thirteenth transmembrane helix seen in the mammalian crystals but not in the chicken.
Spatial organization of the protein domains of the intermembrane region.
Figure 3c shows a slab including the extrinsic domains in the inter-membrane region. The two cytochrome c1 molecules(purple) interact with each other through loops which surround an empty area around the two-fold axis. The hinge protein (light blue) and the external ends of subunits 7 (dark blue) and 10 (green) interact with cytochrome c1 on the side away from the dimer interface.
The ironsulfur protein has no contacts with the other extrinsic domains in the chicken crystals, but the ironsulfur cluster is close to the heme of cytochrome c1 of the opposite monomer, that is, the cytochrome c1 with which its transmembrane helix is not associated. Thus assignment of the Rieske protein to monomers is not obvious in this region. Taking monomers to be as defined in the previous section, the ironsulfur cluster is in a position to interact with cytochromes b and c1 of the other monomer.
The ironsulfur protein position varies slightly (up to 4.5 Å) in
different native crystal forms, as discussed below and reflected in the
inter-center distances in Table 2. In the two beef crystals the ironsulfur
protein is closer to cytochrome c1 than shown for the chicken crystal in Figure
3c, and there is contact between their electron density contoured at 2.0
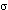 or below. This contact will be described in a later section.
or below. This contact will be described in a later section.
Orientation and environment of the hemes of cytochrome b and c1.
Hemes consist of iron ligated by protoporphyrin IX. The four corners of the porphyrin ring system are formed by 4 pyrrole rings, labeled A - D in Figure 1. Cytochromes contain protoporphyrin IX in which rings A and D are substituted with methyl and propionic side chains, while rings B and C are substituted with vinyl and methyl side chains. Cytochromes b contain B-type hemes which are protoporphyrin IX attached to the protein only by the axial ligand(s), while C-type hemes are further attached by addition of cysteine SH groups across the double bonds of the vinyl groups.
In our final averaged and extended experimental phases the electron density was clear enough (Figures 1 and 4) to recognize the plane of the porphyrin rings and locate the density due to the propionate side chains. This allows orientation of the hemes, with only the ambiguity (in the case of the b hemes) of a possible 180 degree rotation about the axis between heme atoms CHA and CHC relating porphyrin rings A and B to D and C (this results only in exchange of vinyl and methyl groups which cannot be reliably distinguished from the density in these maps).
In the case of cytochrome c1 the heme was assumed to have the same orientation as mitochondrial cytochrome c relative to the features conserved between the two cytochromes (see below). This located the propionates on the heme corners which had density for propionate, although the dihedral angles had to be adjusted to fit the density.
In our structure the planes of the porphyrin rings of both b hemes are roughly parallel to the direction of the helices, i.e. perpendicular to the plane of the membrane. A vector between two opposing pyrrole rings (e.g. B and D) is roughly parallel to the helix axes. For both hemes a propionate-bearing pyrrole ring faces outward to the surface of the membrane, while a methyl- and vinyl-bearing ring faces toward the center.
We have modeled both hemes with the D ring facing outward and the B ring facing
the center. Figure 4a shows the high potential b heme viewed edge on, with the
axis of the 4-helix bundle running vertically. The D propionate, in front
near the viewer, reaches downward toward the membrane surface on the matrix
side, with the carboxylate oxygens in hydrogen bonding distance of 3 residues:
N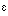 of W32, Nd of N207, and O
of W32, Nd of N207, and O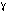 of S107. The other propionate
side chain, from the lower back corner of the heme, bends around toward the
viewer and forms an ion pair with the side chain of R101 (This arrangement of
the propionate differs from that in the structure of Xia et al.). This
propionate and R101 form an extremely strong tube of density connecting the
heme to helix B, which is unbroken even when contoured at 3.5 . The
Nd of the heme axial ligand H98 may be hydrogen bonding with the carbonyl
oxygen of the same residue (H98).
of S107. The other propionate
side chain, from the lower back corner of the heme, bends around toward the
viewer and forms an ion pair with the side chain of R101 (This arrangement of
the propionate differs from that in the structure of Xia et al.). This
propionate and R101 form an extremely strong tube of density connecting the
heme to helix B, which is unbroken even when contoured at 3.5 . The
Nd of the heme axial ligand H98 may be hydrogen bonding with the carbonyl
oxygen of the same residue (H98).
On the low-potential b heme (Figure 4b) the propionates come within hydrogen-bonding distance of nitrogens in the side chain of R81, but there is no density connection at 1.5 sigma contour level. The density for the imidazole rings of the heme axial ligands H84 and H183 is fairly flat and seems to indicate that both rings lie in a plane passing through the nitrogen atoms of pyrrole rings A and C of the porphyrin. Thus asymmetry of the axial ligands is probably not the cause of the unusual EPR properties (27) of the low-potential cytochrome b heme.
There seems to be a hydrogen bond from the N of H84 to O of T48 and
perhaps from Nd of H183 to the backbone amino group of G131 or carbonyl oxygen
of V130. As seen in Figure 4b, these interactions result in the electron
density of the imidazole rings being connected almost equally to the helix
bearing the histidine (helix B or D) and its neighbor (helix A or D). The plane
of the heme is nearly perpendicular to lines between the B and C helices and
the A and D helices, rather than a line between the histidine bearing helices B
and D.
of H84 to O of T48 and
perhaps from Nd of H183 to the backbone amino group of G131 or carbonyl oxygen
of V130. As seen in Figure 4b, these interactions result in the electron
density of the imidazole rings being connected almost equally to the helix
bearing the histidine (helix B or D) and its neighbor (helix A or D). The plane
of the heme is nearly perpendicular to lines between the B and C helices and
the A and D helices, rather than a line between the histidine bearing helices B
and D.
The heme ring density (contoured at 2.0 sigma) is in contact with helix A at
G49. It also contacts helix C at G131 if the contour level is reduced to 1.4
. These are highly conserved glycines, presumably because there is no
room for a side chain with the heme in this position. Force exerted on the heme
tetrapyrrole ring by contact with the helices may be responsible for the
unusual EPR properties of the low-potential cytochrome b heme (27, 28).
Inhibitor binding sites.
The inhibitors antimycin, myxothiazol, and stigmatellin, when present in stoichiometric excess during crystallization, resulted in electron density increases that could be interpreted as due to the bound inhibitors. The general position of the antimycin and myxothiazol binding sites is similar to that inferred from the Figures of Xia and coworkers in references 9 and 10. Although the limited resolution does not allow detailed atomic model-building, we have constructed speculative models consistent with the electron density. Other models may also be consistent, so the orientation and configuration depicted should not be taken as definitive. These inhibitor binding sites, and especially the Qo site, are the target of active drug design efforts to produce environmentally safe and effective plant protection fungicides for agricultural use (29-31). Details of the binding sites correlated with site-directed and inhibitor-resistant mutation data will be described in a separate report after improved phases from a refined structure of the protein become available.
The antimycin site. The putative antimycin site was near the high
potential heme of cytochrome b, in a cavity surrounded by the heme, the
transmembrane helices A, D and E, and the amphipathic
surface helix a. In Figure 5a the viewer is at the position of helix
A, which is invisible in this slab. In our model the aromatic
formamido-salicylate ring of antimycin interacts with the heme propionate and
residues in helices A, D, and a. We have modeled it with
the phenolic OH and formamido groups directed toward the protein of helix
A and the aliphatic atoms on the other side of the ring towards the heme
propionate A side chain. This close approach of the aromatic ring to the heme
was expected from the effect of antimycin on the alpha absorption peak of the
high potential heme and the fluorescence quenching of antimycin when
specifically bound at this site (32). The side chain ring of residue F221
closely approaches one face of the salicylate ring. Aspartate 21 in the
amphipathic helix a apparently hydrogen-bonds with some oxygen in the
9-membered saturated ring of antimycin. The 3 - 6 carbon aliphatic side-chain
also has no interactions and is somewhat disordered, we see density only for
the first 3 carbons. The isobutyryl side chain of antimycin may hydrogen bond
with O of T194 in the chicken complex, however this residue is not
conserved among antimycin-sensitive species. Residues N33 and D229 also appear
to have interactions with antimycin, not visible in Figure 5A
The stigmatellin site. The stigmatellin binding pocket (Figure 5b) is formed by the C-terminak end of helix C, the cd1 helix, the ef linker including the highly conserved -PEWY- sequence and the ef helix, and the N-terminal end of helix F. There are density connections at 1.5 sigma between stigmatellin and residues P271, F275, and M125 of cytochrome b, and H161 of the Rieske protein which has moved from its position in the native crystal.
There is one mass of density the right size for the head group of stigmatellin, suspended between H161 of the Rieske protein and P271 of cytochrome b. We have modeled H161 hydrogen-bonding with methoxy and keto oxygens on one side of the aromatic ring system as proposed(19, 20) based on the structure (33, 34) of stigmatellin bound at the Qb site of bacterial photosynthetic reaction center. P271 is interacting with a methoxy on the other side of the stigmatellin head group, and F275 and M125 are interacting with the hydrophobic tail of the inhibitor. Residues 126-129 of helix C, and 140-147 of the cd linker, are close but not connected at this density contour level. In the native crystals Y279 passes through the region where we have modeled the stigmatellin head group, but in the stigmatellin crystal Y279 has moved and is interacting with R283 and with the Rieske backbone around C160.
The myxothiazol site. Myxothiazol (not shown) binds in roughly the same place as stigmatellin, but displaced a little toward the center of the membrane and toward the low potential b heme. It also makes density contact with P271, but where stigmatellin reaches outward from p271 toward the Rieske protein, myxothiazol and MOA-stilbene reach toward Y132 and F129 in the C helix, in the vicinity of the low potential heme. As with stigmatellin, crystals with these inhibitors show increased density around F275 and M125.
In the absence of any inhibitor the myxothiazol binding pocket contains some density around F275 and M125 not accounted for by the protein model. This may be due to endogenous ubiquinone present at fractional occupancy.
Structure and location of the Rieske ironsulfur protein.
Electron density in the globular extrinsic domain of the Rieske ironsulfur
protein is weaker than in the rest of the structure. The anomalous signal from
the iron-sulfur cluster was weaker than that of the cytochromes, despite the
presence of two irons in the cluster. The backbone density is connected only
when contoured at 1 (Figure 6) or lower, whereas the cytochrome b
backbone was continuous when contoured at 3 . However the density was
good enough to unambiguously locate the known structure of the Rieske extrinsic
domain (14). Figure 6 shows correlation of the electron density with a model
constructed by rotating and translating the published structure to best fit the
density. The protein backbone is nearly all covered with electron density at a
1 contour level, and side chain density is present for many residues
at this level. It can be hoped that when improved phases are available from a
refined model (using omit map techniques to avoid model bias) the side chain
interactions of the Rieske with the rest of the complex may be studied in some
detail.
As predicted from hydropathy plots (35, 36) and molecular engineering results (35), the ironsulfur protein has a membrane-spanning helical segment near the N-terminus. This was removed before residue 68 by proteolysis in preparing the soluble form for structure determination (14), and the residues 68 and 69 were disordered in the crystal, so residue 70 is the first present in the structure used for the model of Figure 6a. However the electron density in our map continues where the model stops (Figure 6a) and connects to a transmembrane helix. The transmembrane helix is well ordered and sequence could be assigned without difficulty.
The N-terminal 24 residues are on the matrix side, and interact with subunit 1
(not shown). Residues 25 to 62 form a transmembrane helix, in close proximity
with the transmembrane helices of subunit 10 and cytochrome c1. The
transmembrane helix is slightly curved and highly slanted. It passes through
the membrane at an angle of about 32deg. to the 2-fold axis, assumed
perpendicular to the membrane. This high degree of tilt accounts for the
length of the transmembrane helix (37 residues), which had led to suggestions
of 2 transmembrane helices for the Rieske protein(36). At the end of the
helix, residues 60 - 66 are locked in a tight vise provided by residues from
both cytochrome b monomers. In monomer A (whose transmembrane domain includes
the transmembrane helix of the Rieske protein E) residues 55 and 73 - 76 from
the turn between the ab helix and helix B contribute to the vise
with density connection to the Rieske protein at 2.5 . In monomer B,
(with which the head interacts), residues 163-169, from the cd2 helix,
and 178 from the N-terminal end of helix D contribute the other face of the
vise with density connection to the Rieske protein at 1.7 .
Residues 67 - 73 are more extended and provide a flexible "tether" connecting the extrinsic domain of the Rieske protein to the transmembrane helix. Residues 70-73 of the published model do not quite fit the electron density and will probably change as the model is refined. It is not surprising that the conformation of these residues changes when the transmembrane helix is cleaved in producing the soluble form, especially since residues 68 and 69 are disordered in the structure of the soluble domains14).
Figure 6b shows a close-up of the ironsulfur cluster region of the Rieske
protein. The two histidine ligands, residues 141 and 161, are clearly evident
as bulges in the density at the tip of the protein. The individual iron atoms
are not resolved at this resolution and temperature factor, but form a
lozenge-shaped mass when contoured at 5 (orange map).
As described above, the ironsulfur cluster of one monomer is in a position to interact with cytochromes b and c1 of the other monomer. The globular extrinsic domain of the ironsulfur protein is oriented with its beta sheets running roughly perpendicular to the surface of cytochrome b. The turns involving residues 70, 92, 176-184, 142, and 196 are facing toward cytochrome b. However the approach is not close. Except for the transmembrane helix, only residues 141-143 (one of the turns which encloses the cluster) makes electron density contact with cytochrome b. This contact, depicted in Figure 7, seems to involve hydrophobic interaction of L142 with T265 of cytochrome b and the carbonyl oxygen of G143 with that of L263. This interaction stabilizes the position of the Rieske protein in our native crystals, with the cluster close to the heme of cytochrome c1 but far from the putative quinol oxidation site Qo.
The other loop enclosing the cluster (residues around 161) faces toward
cytochrome c1, approaching the heme propionates and residues 106 and 145 of
cytochrome c1. There is however no contact with cytochrome c1 at 1.0
in the chicken crystals. The small number of contacts with the rest of the
dimer probably accounts for the high B-factor of the Rieske extrinsic domain,
and suggests that the domain is mobile. This mobility is somewhat restricted
in one monomer of the chicken crystals and in the beef and rabbit hexagonal
crystals by inter-dimeric crystal contacts involving the extrinsic domain of
the Rieske protein.
A dramatic conformational change involving the Rieske protein.
Based on published distances between iron centers, the ironsulfur protein in the tetragonal beef crystals(12, 13) is in a markedly different position than it is in any of our native crystals. When the chicken cytochrome bc1 complex was treated with a saturating amount of stigmatellin before crystallization, we found the ironsulfur protein in a conformation (Figures 5b and 8) similar to that reported for the tetragonal beef crystals (13). The relative positions of the redox cofactors in chicken crystals in the presence and absence of stigmatellin are shown schematically in Figure 8, together with distances between cofactors. The distances from the ironsulfur cluster to the heme iron peaks of cytochrome c1 and the low potential cytochrome b heme are listed in Table 2 for all our crystals and for the tetragonal beef crystals.
The two conformations of the Rieske protein in chicken crystals are compared by superposition in Figure 9. A stereo view of both conformations together with cytochrome b and the heme of cytochrome c1 is shown in Figure 9a. Figure 9b shows the whole dimer in light gray, with the two conformations of the Rieske protein as well as stigmatellin and the heme of cytochrome c1 highlighted with color. Figure 9c shows a close-up of the Rieske soluble domain in the whole dimer. In the presence of bound stigmatellin, the cluster has moved down along the surface of cytochrome b and has a strong density connection (1.6 sigma) to density which we assign to stigmatellin because of its absence in the native crystals, as mentioned in the description of the stigmatellin binding site. This movement is accomplished by a rotation of 57deg. about an axis passing through the ironsulfur protein near residues 93 and 182.
The transmembrane helix and matrix-side portion are unchanged in the presence of stigmatellin. The coil consisting of residues 68-73 is stretched out in the presence of stigmatellin, allowing this end of the soluble domain to move farther from the membrane as the Fe2S2 cluster on the other end moves closer. In a crystal with both stigmatellin and antimycin bound, the ironsulfur position was essentially the same as that with only stigmatellin. In crystals with only antimycin or myxothiazol bound the ironsulfur position was similar to that in the crystals without inhibitor.
Folding pattern of cytochrome c1 compared to various c cytochromes.
The cytochrome c1 subunit forms a wedge-like structure containing the heme in
the extrinsic domain, with a C-terminal membrane anchor extending up alongside
cytochrome b, parallel to helix E. Figure 10 compares the backbone
folding pattern of cytochrome c1 and mitochondrial cytochrome c, a member of
Ambler's type I cytochromes c. Type I cytochromes c have 5 helical segments,
labeled  1 - 5 in Figure 10. The three helices (1,
3, and 5) which have more than 2 turns are conserved in
cytochrome c1 and occupy the same positions relative to each other and to the
heme. They are colored alike and labeled on both molecules in Figure 10.
Conserved aromatic residues involved in interaction between helix 1 and helix 5
(F10 and Y97 in mitochondrial cytochrome) are present as Y33 and F189,
respectively. The tripeptide starting at residue 30 in mitochondrial
cytochrome c is conserved -PNL-, with the proline carbonyl supplying a hydrogen
bond to Nd of the histidine heme ligand and the lysine providing
hydrophobic environment for the heme ring. This aligns with cytochrome c1 -PDL-
starting at residue 111, which is conserved in all cytochromes c1 except that
of Rhodobacter sphaeroides, which barring a sequencing error has ADL.
These similarities justify inclusion of cytochrome c1 in the family of type I
cytochromes. A detailed comparison and structure-based alignment of cytochrome
c1 with mitochondrial cytochromes c, bacterial cytochromes c2, and the "small"
type I cytochromes c6 and Pseudomonas aeruginosa cytochrome c will be reported
separately.
1 - 5 in Figure 10. The three helices (1,
3, and 5) which have more than 2 turns are conserved in
cytochrome c1 and occupy the same positions relative to each other and to the
heme. They are colored alike and labeled on both molecules in Figure 10.
Conserved aromatic residues involved in interaction between helix 1 and helix 5
(F10 and Y97 in mitochondrial cytochrome) are present as Y33 and F189,
respectively. The tripeptide starting at residue 30 in mitochondrial
cytochrome c is conserved -PNL-, with the proline carbonyl supplying a hydrogen
bond to Nd of the histidine heme ligand and the lysine providing
hydrophobic environment for the heme ring. This aligns with cytochrome c1 -PDL-
starting at residue 111, which is conserved in all cytochromes c1 except that
of Rhodobacter sphaeroides, which barring a sequencing error has ADL.
These similarities justify inclusion of cytochrome c1 in the family of type I
cytochromes. A detailed comparison and structure-based alignment of cytochrome
c1 with mitochondrial cytochromes c, bacterial cytochromes c2, and the "small"
type I cytochromes c6 and Pseudomonas aeruginosa cytochrome c will be reported
separately.
The heme pocket and exposure at the C corner. Mitochondrial cytochromes c have the C corner of the heme exposed at the "front" face, where electron transfer is believed to take place. This corner is also exposed in cytochrome c1. The exposed C corner of the heme is surrounded by 3 stretches of protein, consisting of residues 36-41 (corresponding to cytochrome c 10-18), 111-114 (corresponding to cytochrome c 30 - 33 and containing the heme-bracing proline mentioned above), and 150-163 (corresponding to cytochrome c 70-83).
Major differences between cytochromes c and c1 are the result of additions or
deletions in loop regions. Bovine cytochrome c1 has an N-terminal extension of
24 residues before helix 1, compared to 1 to 11 residues in
mitochondrial cytochromes c. This has no defined secondary structure. The
first 13 residues run parallel to the last helix of subunit 8, the acidic hinge
protein. This forms the main interface of cytochrome c1 with the hinge protein,
supplemented on it's "1" end by residues 132-140 and on its "13" end by
residues 176-179. In P. denitrificans, which has no hinge protein,
there is a 237-residue acidic insertion in this region which may serve the
function of, or even be the evolutionary forerunner of, the hinge protein.
After helix 1 and the "fingerprint" CXYCH heme-binding stretch, which
are similar in the two cytochromes, cytochrome c1 has a long insertion
(residues 52 - 109) between residues 28 and 29 of cytochrome c. This expands
the loop consisting of residues 19-28 in cytochrome c into a long, branched
loop structure. The first branch folds back and contacts residues around the
beginning of helix 1. An acidic stretch (residues 58 - 77) including
the "Y" where the two branches come together, has been implicated in binding
cytochrome c (37). The second branch of the insertion, including residues
68-85, reaches across the dimer to bring residue G78 into contact with E99 of
cytochrome c1 in the other monomer. Thus this insertion includes both sides of
a dimer contact. Residues 98-106 form the 3-turn alpha helix 2' which
has no counterpart in cytochrome c. The bacterial cytochromes c1 (Rhodobacter
and Paracoccus) have a 10-residue deletion in this area relative to the bovine
cytochrome, so this helix may be absent in the bacterial cytochromes.
The loop between the conserved tripeptide 30:PNL/111:PDL and helix 3 (residues 34-59 in cytochrome c, containing helix 2) is greatly shortened in cytochrome c1 by deletion of 18 residues corresponding to cytochrome c residues 41-58. This stretch in cytochrome c covers the heme propionates and insulates the heme from the aqueous environment. It is also deleted in the so-called "small" type 1 cytochromes such as the P. aeruginosa cytochrome c (Protein database entries 351C, 451C) and chloroplast cytochrome c6 (PDB entries 1CYI, 1CYJ). Interestingly, the bacterial cytochromes c1 have a 17-residue insertion here relative to bovine cytochrome c1, suggesting that this loop may be present. The exposed propionates in chicken or bovine cytochrome c1 are directed toward the Rieske ironsulfur cluster, so it would seem this loop has been removed to open a second path for electron entry to the heme from the Rieske protein. If the bacterial enzymes also utilize this pathway, then the loop corresponding to cytochrome c residues 41-58 must be in a completely different position than in cytochrome c.
The stretch between helix 3 and the methionine heme ligand, which
consists of helix 4 in cytochrome c, is longer by 17 residues in
bovine cytochrome c1, and the length varies between vertebrate, yeast and
bacterial cytochromes c1. There is no helix corresponding to 4.
Residues 132 - 140 in this insertion are involved in the interface with the
"hinge" protein.
Between the methionine heme ligand and helix 5 there are 6 residues in cytochrome c but 18 in the c1 cytochromes. This region is relatively acidic and has one conserved aromatic residue, 171. This region has been implicated in cytochrome c binding (38). It contributes to the interface with the "hinge" protein.
After helix 5 is interrupted by a turn at P197, the transmembrane
helix 6' starts immediately at residue 198. conserved R201 seems to
interact with conserved E195 and with P248 at the end of helix E of cytochrome
b. The TMH of cytochrome c1 is further connected to helix E by density
connections at the positions of residues G205, M208, and M212 connecting with
cytochrome b residues F246, S242, and S238.
A second exposure of the heme on the A-D edge, and the interface with the Rieske protein.
As mentioned above, deletion of the long loop corresponding to residues
41-58 in Tuna cytochrome c results in exposure of the heme propionates to the
surface and to the Rieske ironsulfur cluster, apparently allowing electron
transfer from the cluster to cytochrome c1 at this edge. As shown in Table 2,
the Rieske protein is closer to cytochrome c1 in the two beef crystals. These
very likely represent the configuration during electron transfer from the
ironsulfur cluster to the cytochrome. Figure 11 shows the Rieske protein -
cytochrome c1 interface in the beef hexagonal crystals. H161 of the Rieske
protein, which provides one of the ligands to the Fe2S2 cluster, is 4.5
Å from the D propionate oxygen atom and 9. Å from the edge of the
heme  -bonded system at the C3D atom. There is actually electron density
contact between the propionate and H161 at a contour level of 1.0 .
Even when contoured at 2 sigma as in Figure 11 there is density contact between
the Rieske protein around C160 and cytochrome c1 around G107. C160 is one of
the cysteines in the cluster loops of the Rieske protein, which form a
disulfide bond holding the cluster-binding loops together. In cytochrome c1,
G107 is between helix 2' and the heme-bracing proline P111. The other
heme propionate, propionate A, is farther from the Rieske cluster and is within
hydrogen-bonding distance of R120 and Y126 of cytochrome c1.
-bonded system at the C3D atom. There is actually electron density
contact between the propionate and H161 at a contour level of 1.0 .
Even when contoured at 2 sigma as in Figure 11 there is density contact between
the Rieske protein around C160 and cytochrome c1 around G107. C160 is one of
the cysteines in the cluster loops of the Rieske protein, which form a
disulfide bond holding the cluster-binding loops together. In cytochrome c1,
G107 is between helix 2' and the heme-bracing proline P111. The other
heme propionate, propionate A, is farther from the Rieske cluster and is within
hydrogen-bonding distance of R120 and Y126 of cytochrome c1.
Conserved residue A119 of cytochrome c1 interacts with the ef linker of cytochrome b around residue P254. The contact described in Figure 7 between the Rieske protein and the ef linker at residues 263 and 265 is still present. This stretch of cytochrome b (254 - 265) linking the Rieske protein to cytochrome c1 is supported by the angle between helices C and cd1 at residue P135.
Figure 12 shows a slab through the plane containing the Rieske ironsulfur cluster, the heme of cytochrome c1, and the probable site for the heme of cytochrome c bound to cytochrome c1. The position of the ironsulfur protein is that from the chicken crystals, in the beef crystals it is about 4 Å closer to cytochrome c1. This diagram illustrates the possibility of electron transfer into cytochrome c1 via the D propionate and out of cytochrome c1 via the C corner of the heme to cytochrome c.
METHODS
Purification and crystallization. A detailed report on purification and crystallization is being prepared for publication. Briefly, the cytochrome bc1 complex was purified from different vertebrate heart tissues essentially as described for the potato complex (39). Mitochondria were prepared by the method of Smith(40) and solubilized using the detergent dodecyl maltoside. The complex was isolated from the extract by chromatography on DEAE Sepharose CL6B and further purified by size exclusion chromatography on Sepharose CL6B. The protein was concentrated to around 200 uM by ultrafiltration through an Amicon YM-100 membrane, precrystallized by mixing with 100 mM KMES pH 6.5, + 10% PEG-4000, and redissolved in 20 mM K-MOPS 7.5, 100 mM NaCl. Aliquots (5-20 ul) were mixed with an equal volume of precipitant containing 100 mM KMES pH 6.7 and subjected to vapor diffusion against 25% glycerol.
To co-crystallize the bc1 complex with the high-affinity inhibitors antimycin, myxothiazol, or stigmatellin, the inhibitor was added from an ethanolic solution (the final ethanol concentration was below 1% v:v) in a 1.5 - 2.0-fold molar ratio to the pooled fractions from the final column at a protein concentration of 5-10 uM before concentrating and precrystallizing as above.
Cryogenic data collection for chicken bc1 crystals. After crystallization was complete (5-30 days after setup) 20 ul of cryoprotectant containing 10 mM K-MES pH 6.7, 10 mM OG, 25% glycerol, 10% PEG 4000k was added and the reservoir was changed to 35% glycerol for further concentration of glycerol and PEG without increasing ionic strength. After this equilibration or in some cases after further soaking in cryoprotectant consisting of 30% glycerol, KMES, and OG, crystals were frozen in liquid ethane or nitrogen, or directly in the cryogenic stream,. and data were collected at 70deg. - 100deg. K.
A suitable procedure for cryogenic data collection of the beef and rabbit crystals has not yet been developed. Data sets were obtained by merging data from multiple crystals and from different areas of crystals larger than the x-ray beam. Generally no more than 10 exposures of the beef hexagonal bipyramids could be collected at a synchrotron source before diffraction decayed and the crystal had to be translated or replaced. The monoclinic beef crystals seemed somewhat more resistant to radiation damage; however a complete data set could not be collected from a single crystal.
Data Reduction and crystallographic computing. Diffraction patterns collected on image plates were indexed and spot intensity integrated using the programs DENZO(41). Intensities from different diffraction patterns were scaled together with SCALEPACK(41). Heavy atom derivatives were analyzed using XtalView (42). The CCP4 package(43) was used for final heavy atom refinement and phase calculation (MLPHARE), for finding molecular replacement solutions (programs ALMN and TFFC) and for routine calculations such as scaling, map calculation, and back transformation. The RAVE package(44) was used for molecular averaging, map skewing, and rotation-translation operator improvement. Model building and examination was with the program O (45), which was also used to make Figures 1, 4-6, and 11. The ribbon diagrams in Figures 2, 3, 8, and 10 were prepared using Molscript (46) and rendered with Raster3D(47). Models for the inhibitors were constructed with Quanta (Molecular Simulation Inc.) and fitted to the density using the program O.
Phasing. The chicken crystals were phased by isomorphous replacement and the resulting electron density was used to phase the other crystal forms by molecular replacement. The phases were improved and extended to the resolution limit of the data by multi-crystal and noncrystallographic symmetry averaging. Details are presented in Table 3.
Location of iron centers from anomalous data. Anomalous data at wavelength near the iron K absorption edge were collected at beamline 1- 5AD at the Stanford Synchrotron Radiation Laboratory. An EXAFS scan of a native chicken crystal was used to select 4 wavelengths for data collection: 7131, 7111.2, 7749, and 12300 eV (1.739, 1.7435, 1.600, and 1.008 Å). To locate iron centers, a Bivoet difference map was made using the data at 7131 eV amplitudes and isomorphous phases improved by averaging. Iron centers were located as peaks in the map.
Location of the ironsulfur protein structure in the electron density. The extrinsic domain of the Rieske ironsulfur protein has relatively weak electron density. Instead of building a model for this region, we used the published structure of the extrinsic domain of the Rieske protein solved from crystals of a proteolytic fragment(14) after rotation and translation to the orientation and position in our crystals, determined as follows.
By putting the ironsulfur cluster of the model at the position of the ironsulfur peak in the Bivoet difference map and rotating about this point to maximize overlap of the structure with electron density, we were able to approximately orient the published structure by eye. The orientation was refined by optimizing correlation between our electron density and a density map calculated from the published structure. For this the "improve" option of the Mave program of the Rave averaging package(44) was used to optimize the rotation-translation operator relating the two maps. For the other crystal forms and for the chicken crystals with stigmatellin bound, the orientation of the Rieske protein was different so the process was repeated (because anomalous data was not available, a peak in the electron density map was used for the initial cluster location ). In difficult cases a small mask around the cluster was used for the initial optimization, to improve the radius of convergence. This mask was gradually increased to include the whole extrinsic domain of the ironsulfur protein for more accurate orientation.
Map calculation. The maps presented here are calculated using
coefficients of (2Fo-Fc) e-i c, where the Fo are from the
experimentally determined intensities and the Fc and c are calculated
from the previous map of the averaging procedure described in Table 3, skewed
into the appropriate cell using the appropriate rotation-translation operators
for each domain. In the case of unobserved reflections, Fo was replaced by Fc
as recommended(48), resulting in coefficients of Fc e-ic for
those terms. This "fill-in" procedure was used both during averaging and in
making the final maps used in the figures. Thus the maps in the figures have
not been averaged. They are made using phases from averaged maps, but use of
2Fo-Fc coefficients reduces bias from the phases. For the figures, Fc and
c are from the final map of the averaging procedure.
c, where the Fo are from the
experimentally determined intensities and the Fc and c are calculated
from the previous map of the averaging procedure described in Table 3, skewed
into the appropriate cell using the appropriate rotation-translation operators
for each domain. In the case of unobserved reflections, Fo was replaced by Fc
as recommended(48), resulting in coefficients of Fc e-ic for
those terms. This "fill-in" procedure was used both during averaging and in
making the final maps used in the figures. Thus the maps in the figures have
not been averaged. They are made using phases from averaged maps, but use of
2Fo-Fc coefficients reduces bias from the phases. For the figures, Fc and
c are from the final map of the averaging procedure.
With the exception of Figure 11, the final map was single-domain averaged over
noncrystallographic symmetry and multiple chicken bc1 crystals, all of the same
space group but with slightly varying cell parameters. The averaged density was
positioned in the cell of the chicken crystal labeled chc01 in Table 3a for
calculating Fc and c, and the Fo values are from this crystal. While
data to 3.1 Å was included, the reflections were very weak in the highest
resolution shells. The R-merge value exceeded 0.25 at resolution beyond 3.5
Å. Omission of the data beyond 3.5 Å has no significant effect on
the appearance of the maps in the figures and the maps from which the model was
constructed, so we consider this to be a 3.5 A structure.
In the case of Figure 11 the observed data Fo were from the hexagonal beef
dataset, and Fc and c values are from 7-crystal 2-domain averaging using
4 different crystal forms. The highest resolution in the beef dataset was 4.5
Å. The fill-in procedure was omitted in making the map for Figure 11 to
avoid including high resolution information from the chicken crystals.
References
1. Mitchell, P. (1961) Coupling of phosphorylation to electron and proton transfer by a chemi-osmotic type of mechanism. Nature (London) 191, 144-148
2. Hatefi, Y, Galante, Y.M., Stiggall D.L., Ragan C.I. Proteins, polypeptides, prosthetic groups, and enzymic properties of complexes I, II, III, IV, and V of the mitochondrial oxidative phosphorylation system. Methods Enzymol 56, 577-602 (1979)
3.. Hinkle, P.C., Kumar, M.A., Resetar ,A., and Harris ,D.L. Mechanistic Stoichiometry of Mitochondrial Oxidative Phosphorylation. Biochemistry 30, 3576-3582. (1991)
4. Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor... Biol. 62, 327-367 (1976)
5. Crofts, A.R. The mechanism of Ubiquinol:cytochrome c Oxidoreductases of Mitochondria and of Rhodopseudomonas sphaeroides. In: The Enzymes of Biological Membranes,, Vol. 4 (ed Martonosi, A.N.) pp. 347-382. (Plenum Publ. Corp., New York. 1985)
6. Schägger H., Link, Th. A., Engel, W.D., and von Jagow, G. Isolation of the Eleven Protein Subunits of the bc1 complex from beef heart. Methods in Enzymology 126, 224-237. (1986)
7. Gencic, S., Schägger H., and von Jagow G. Core-I Protein of Bovine Ubiquinol Cytochrome-c Reductase- an Additional Member of the Mitochondrial-Protein-Processing Family - Cloning of Bovine Core-I and Core-II cDNAs and Primary Structure of the Proteins. European Journal of Biochemistry 199, 123-131. (1991)
8. Weiss, H. and Leonard, K. Structure and Function of Mitochondrial Ubiquinol:Cytochrome c Reductase and NADH:Ubiquinone Reductase. Chemica Scripta 27B, 73-81. (1987)
9. Yue, W.H., Zou, Y.P., Yu, L., and Yu, C.A. Crystallization of mitochondrial ubiquinol-cytochrome c reductase. Biochemistry 30, 2303-2306. (1991)
10. Kubota T., Kawamoto M., Fukuyama K., Shinzawa-Itoh K; Yoshikawa S; Matsubara H. Crystallization and preliminary X-ray crystallographic studies of bovine heart mitochondrial cytochrome bc1 complex. J. Mol. Biol. 221, 379-382. (1991)
11. Berry, E.A. , Huang, L.-s , Earnest, T.N. and Jap, B.K. X-ray Diffraction by Crystals of Beef Heart Ubiquinol:Cytochrome c Oxidoreductase. J. Mol. Biol. 224, 1161-1166 (1992)
12. Yu, C.-A., Xia, J.-Z., Kachurin, A.M., Yu, L., Xia, D., Kim, H. and Deisenhofer, J. Crystallization and preliminary structure of beef heart mitochondrial cytochrome-bc1 complex. Biochim. Biophys. Acta 1275, 47-53. (1996)
13. Xia, D., Yu,C.-A., Kim, H., Jia-Zhi Xia, J.-Z., Anatoly M. Kachurin, A.,M., Zhang, L., Yu, L., Deisenhofer, J. Crystal Structure of the Cytochrome bc1 Complex from Bovine Heart Mitochondria . Science 277, 60-66 (1997)
14. Iwata, S., Saynovits, M., Link, Th.A. and Michel, H. Structure of a water soluble fragment of the 'Rieske' iron-sulfur protein of the bovine heart mitochondrial cytochrome bc1 complex determined by MAD phasing at 1.5Å resolution. Structure 1996, 4: 567-579. (1996)
15. Berry, E.A., Huang, L.-s., Shulmeister, V.M., and Kim, S.-H. A New Form of Crystal of Bovine Heart Ubiquinol:Cytochrome c Oxidoreductase- Determination of Space Group and Unit Cell Parameters. Acta Cryst. D51 235-239. (1995)
16. Meinhardt, S.W. and Crofts, A.R. Kinetic and Thermodynamic Resolution of Cytochrome c1 and Cytochrome c2 from Rps. sphaeroides.. FEBS Lett., 149, 223-227. (1982)
17. Tsai, A.-L., Olson, J.S. and Palmer, G. The oxidation of yeast Complex III. Evidence for a very rapid electron equilibration between cytochrome c1 and the iron-sulfur center. J. Biol. Chem. 258, 2122-2125 (1983)
18. Link, T.A. and Iwata, S. Functional aspects of the structure of the 'Rieske' iron-sulfur protein of bovine heart mitochondrial cytochrome bc1 complex. Biochim. Biophys. Acta 1275, 54-60. (1996)
19. Link, T.A. The role of the "Rieske" iron sulfur protein in the hydroquinone oxidation (Qp-) site of the cytochrome bc1 complex: The "proton-gated affinity change" mechanism. FEBS Letters. 412(2):257-264 (1997)
20. Link, T.A. "Structural aspects of the cytochrome bc1 complex" in: "Frontiers of Cellular Bioenergetics: Molecular Biology, Biochemistry and Physiopathology" (eds S. Papa, F. Guerrieri and J.M. Tager,) pp. xxx-yyy. (Plenum Publishing Co., New York - London. 1998)
21. Gonzalez-Halphen, D., Lindorfer, M.A., and Capaldi, R.A. Subunit Arrangement in Beef Heart Complex III. Biochemistry 27, 7021-7031. (1988)
22. Crofts, A.R., Robinson, H.H., Andrews, K., Van Doren, S. and Berry, E. Catalytic sites for reduction and oxidation of quinones. In "Cytochrome Systems: Molecular Biology and Bioenergetics" (eds Papa, S., Chance, B. and Ernster, L) pp. 617-624 (Plenum Publ., New York. 1987)
23. Crofts, A.R., Hacker, B., Barquera, B., Yun, C.-H. and Gennis, R. Structure and function of the bc-complex of Rhodobacter sphaeroides. Biochim. Biophys. Acta 1101, 162-165 (1992)
24. Degli Esposti, M. et al. "Mitochondrial cytochrome b: evolution and structure of the protein", Biochim. Biophys. Acta 1143, 243-271 (1993)
25. Musatov A, Robinson N.C. Subunit analysis of bovine cytochrome bc1 by reverse-phase HPLC and determination of the subunit molecular masses by electrospray ionization mass spectrometry. Biochemistry 33, 10561-10567 (1994)
26. Brandt U, Yu L, Yu CA, Trumpower BL The mitochondrial targeting presequence of the Rieske iron-sulfur protein is processed in a single step after insertion into the cytochrome bc1 complex in mammals and retained as a subunit in the complex. J Biol Chem 268:8387-8390 (1993)
27. Carter K.R., Tsai A., Palmer G. The coordination environment of mitochondrial cytochromes b. FEBS Lett. 132, 243-246 (1981)
28. Salerno J.C. Cytochrome electron spin resonance line shapes, ligand fields, and components stoichiometry in ubiquinol-cytochrome c oxidoreductase. J. Biol Chem. 259, 2331-2336 (1984)
29. Beautement K., Clough J.M., Defraine P.J., Godfrey C.R.A. Fungicidal Beta-Methoxyacrylates - From Natural Products To Novel Synthetic Agricultural Fungicides. Pesticide Science 31, 499-519. (1991)
30. Clough, J.M., and Godfrey, C.R.A. Growing Hopes. Chemistry In Britain 31,466-469. (1995)
31. Sauter, H., Ammermann, E., and Roehl, F. Strobilurins - From natural products to a new class of fungicides. in: "Crop Protection Agents from Nature" (ed Leonard G. Copping) pp. 50 - 81. (The Royal Society of Chemistry, Thomas Graham House, Cambridge, UK. 1996)
32. Slater E.C. The mechanism of action of the respiratory inhibitor, antimycin. Biochim Biophys Acta 301:129-154 (1973)
33. Lancaster, C.R. and Michel, H. in "Reaction centers of photosynthetic bacteria. Structure and Dynamics". International Workshop Feldafing III (ed Michel-Beyerle, M.E.,) pp. (Springer-Verlag, Berlin. 1995)
34. Lancaster, C.R.D., and Michel, H. The coupling of light-induced electron transfer and proton uptake as derived from crystal structures of reaction centers from Rhodopseudomonas viridis modified at the binding site of the secondary quinone , QB. Structure 5, ??-??. (1997) (not yet available)
35. Van Doren, S.R., Yun, C-H., Crofts, A.R. and Gennis, R. Assembly of the Rieske iron-sulfur subunit of the cytochrome bc1 complex in Escherichia coli and Rhodobacter sphaeroides membranes independent of the cytochrome b and c1 subunits. Biochemistry 32, 628-636 (1993)
36. Link, T.A., Schägger, H. and Von Jagow, G. Structural analysis of the bc1 complex from beef heart mitochondria by the sidedness hydropathy plot and by comparison with other bc complexes. In Cytochrome Systems: Molecular Biology and Bioenergetics (eds Papa, S., Chance, B. and Ernster, L.) pp. 289-301. (Plenum Press, New York 1987)
37. Stonehuerner, J., O,Brien, P., Geren, L., Millett, F., Steidl, J., Yu, L., and Yu, C.-A. Identification of the Binding Site on Cytochrome c1 for Cytochrome c. J. Biol. Chem. 260, 5392-5398. (1985)
38. Broger C., Salardi, S., and Azzi, A. Interaction between Isolated Cytochrome c1 and cytochrome c. Eur. J. Biochim.. 131, 349-352. (1983)
39. Berry, E.A, Huang, L.-s. and DeRose, V. Ubiquinol-Cytochrome c Oxidoreductase of Higher Plants. Isolation and Characterization of the bc1 Complex from Potato Tuber Mitochondria. J. Biol. Chem. 266, 9064-9077. (1991)
40. Smith, A.L. (1967) Preparation, properties, and conditions for assay of mitochondria: slaughterhouse material, small scale. Meth. Enzymol.. 10, 81-86
41. Zbyszek Otwinowski "Oscillation Data Reduction Program", in Proceedings of the CCP4 Study Weekend: "Data Collection and Processing"(eds L. Sawyer, N. Isaacs and S. Bailey) pp 56-62 (SERC Daresbury Laboratory, England, 1993)
42. McRee, D. E. A visual protein crystallographic software system for X11/XView. J. Molecular Graphics 10, 44-46 (1992).
43. CCP4 The SERC (UK) Collaborative Computing Project No. 4 "The CCP4 Suite: Programs for Protein Crystallography". Acta Cryst. D50, 760-763. (1995)
44. Jones, T.A. Proc. CCP4 Study Weekend, Molecular Replacement (eds Dodson,E.J., Glover,S., & Wolf W.. pp. 91-105 (SERC Daresbury Laboratory, UK, 1992)
45. Jones T.A.; Zou J.Y.; Cowan S.W., Kjeldgaard M. Improved Methods For Building Protein Models In Electron Density Maps And The Location Of Errors In These Models. Acta Crystallographica A47, 110-119. (1991)
46. Per J. Kraulis "MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures", Journal of Applied Crystallography 24, 946-950. (1991)
47. Merritt E.A.; Murphy M.E.P. Raster3D Version 2.0 - A Program For Photorealistic Molecular Graphics. Acta Crystallographica D50, 869-873. (1994)
48. Rayment, I. Molecular replacement method at low resolution: optimum strategy and intrinsic limitations as determined by calculations on icosahedral virus models. Acta Cryst. A39, 102-116. (1983)
Acknowledgments- This investigation was supported by NIH and by the Office of Biosciences and Environmental Research, US Department of Energy. The work was partially done at SSRL which is operated by the Department of Energy, Division of Chemical/Material Sciences. The SSRL Biotechnology Program is supported by the National Institutes of Health Biomedical Resource Technology Program, Division of Research Resources. We thank Thomas Link and his co-workers for providing coordinates for the soluble domain of the Rieske ironsulfur protein before their release from the Protein Data Bank, Henry Bellamy for the performing the XAFS scan and for advice and help with MAD data collection, SangJin Hong for preparing coordinate files for the inhibitors, and Liang Tong and Dave Schuller for advice on molecular averaging.
* abbreviations used are: "native"- with no heavy atom reagent of inhibitor
added to the protein; Fo and Fc- structure factor amplitudes calculated from
the diffraction pattern (Fo) or from a map or model (Fc); c, structure
factor phases calculated from a map or atomic model; NCS, noncrystallographic
symmetry.
Tables
Table 1. Parameters of Crystal forms used in the structure determination.
#AU Mono
Protein Space Group Unit Cell cell AU (106 Å3) Å3/Da Dmax1
beef bc1
Hexagonal
bipyramid P6522 217 217 378 90deg. 90deg. 120deg. 12 1 15.41 1.28 5.20 3.8
Monoclinic2 P21 118 178 200 90 106 90 8 2 8.02 1.00 4.16 3.7
Rabbit bc1
Hexagonal
bipyramid P6522 212 212 354 90deg. 90deg. 120deg. 12 1 13.78 1.15 4.60 3.5
Chicken bc1
orthorhombic P212121 176 184 242 90deg. 90deg. 90deg. 4 2 7.84 1.96 4.04 3.0
frozen- 169 181 239 90deg. 90deg. 90deg. " " 7.31 1.83 3.76 2.9
1. AU = asymmetric unit. Mono = monomer. Dmax = d-spacing of highest recorded indexable reflection.
2. The monoclinic beef crystals were mistakenly reported as centered orthorhombic (C2221) in ref. R12
Table 2. Distances between the Rieske ironsulfur cluster and cytochromes c1 and b-low.
distance (Å) from
Fe2S2 cluster to:
crystal heme bL heme c1
beef P4122 (from ref 13) 27. 31.
chicken P212121 (+stigmatellin) 26.4 31.6
chicken P212121 34.3 21.3
beef P6522 34.9 17.2
beef P21 35.1 17.5
rabbit P6522 35.5 19.1
Iron peaks located as peaks in electron density from averaged experimental phases improved and extended by molecular averaging, except for chicken P212121 in the absence of inhibitor, in which case Bivoet difference amplitudes were used with improved experimental phases retarded by 90deg..
Table 3. Structure determination.
(a) Diffraction data, Chicken crystals
Unique Completeness(%) X-Ray1
Native: dmin No.Refln Refl (I> 1 sigma) Rmerge (%) Source
chn21 3.6 279,119 70,363 76.3(58.1) 18.6 RA
chc01 3.1 556,456 123,869 91.6(80.5) 10.2 SSRL
chm 3.01 569,255 141,427 96.6(74.2) 16.2 SSRL
chb 2.95 433,902 131,641 81.7(51.2) 27.8 BNL
Derivative:
PIP* 3.50 292,339 86,221 91.8(76.3) 10.8 SSRL
NSDMA* 3.90 425,028 67,109 99.7(79.0) 12.4 RA
tml02 3.50 203,105 61,380 65.2(53.5) 17.1 RA
tml03 4.30 110,201 38,103 74.4(50.6) 21.6 RA
hpdl 4.00 129,187 48,715 78.4(54.0) 20.2 RA
iridium 3.50 177,303 68,522 71.7(43.5) 13.4 RA
tmlssrl 3.50 160,826 61,128 65.6(43.9) 19.9 SSRL
hpdlssrl 3.15 350,204 92,367 71.6(60.2) 19.3 SSRL
1- RA- Rotating anode. SSRL- Stanford Synchrotron Radiation Laboratory, BL7-1.
BNL- Brookhaven National Laboratory X-12b.
(b) Isomorphous Phase determination
Derivatives Rderi No. of Rc Phasing power in Resolution shell (A)
(%) sites 12.56 8.77 6.74 5.47 4.61 3.98 3.50 Total
PIP 13.2 16 0.83 0.88 1.01 1.12 1.12 0.94 0.97 0.86 0.97
NSDMA* 15.2 16 0.88 0.68 0.61 0.76 0.96 0.93 0.96 0.97 0.89
tml02 21.5 23 0.79 1.92 1.81 1.94 1.76 1.24 1.05 0.99 1.32
tml03 28.1 23 0.71 1.84 1.67 1.88 1.67 1.38 - - 1.66
HPDL* 15.3 4 0.89 1.04 0.94 0.98 0.96 0.70 0.64 - 0.82
iridium 20.6 11 0.93 0.49 0.50 0.66 0.84 0.72 0.64 0.66 0.67
Figure of Merit 0.25 0.58 0.62 0.56 0.48 0.41 0.29 0.46
*PIP -
*HPDL - hexphenyl dilead
*NSDMA - N-(5-nitrosalicyl)-(S-decylmercuri)6-aminothiophenol, a putative antimycin analog
The chicken crystals were phased by isomorphous replacement using the 5 crystals derivatized with 4 heavy atom reagents described above. The handedness of the solution was determined by the sign of peaks in the iron Bivoet difference map. The resulting phases were improved by solvent flattening and NCS-averaging using the dimer molecular envelope and NCS operator determined from the heavy atom positions and isomorphous map. The improved phases were used to make a better map which was used as a model for molecular replacement to phase the other crystals . This was achieved by using the RAVE package(8) to make a mask around the bc1 dimer in the chicken cell and translate the density within this mask into a cubic cell 300 Å on each edge, with P1 symmetry. The CCP4 package (ref 8, sfall and almn) was used to calculate structure factors from this P1 map and evaluate the cross-rotation function between these structure factors and the observed structure factor amplitudes of each unphased crystal. The Rave package was again used to rotate the density of the dimer to the chosen rotation solution for each unphased crystal and place it in a P1 cell of the same dimensions as the cell of that crystal. Then the ccp4 package (fft, cad, tffc) was used to calculate structure factors and evaluate the translation function of the oriented model with the unphased crystal. In the case of the hexagonal crystals (beef and rabbit) this was done assuming either p6122 or p6522 symmetry. A strong peak in the translation function was obtained only with p6522 symmetry, so this was assumed to be the correct enantiomer.
The phases were improved by multicrystal and ncs density averaging over 4 crystal forms(Table 1), two of which have 2-fold noncrystallographic symmetry, using the RAVE package. In each cycle unobserved Fo data were replaced by Fcal from the previous map as recommended by Rayment(11) in calculating the new (2Fo-Fc) map. No positivity constraint was placed on the averaged density. Density outside the masks (in the solvent region) was set to a constant value to make the average density of the unit cell zero. In most cases seven crystals were averaged- 4 good but slightly non-isomorphous chicken crystals and one each of beef hexagonal, rabbit, and beef p21 crystals The beef P21 crystal was averaged with a weight of 2 to account for its 2-fold NCS. The chicken crystals were given weight 1.0 each despite the NCS, as the chicken structre was felt to be over-represented. Resolution was limited to 4.0 Å at this stage.
Multi-crystal averaging was achieved by averaging the asymmetric units of each crystal into an imaginary reference cell in which the dimer 2-fold is along the z axis and averaging with similarly repositioned density from each other cell. This not only allowed averaging of crystals of different space groups, but also allowed accurate averaging of "non-isomorphous" crystals of the same space group but different cell parameters. This reference cell was also used for superpositioning density maps from different crystal forms or in the presence and absence of inhibitors for comparison.
Map inspection showed that the ironsulfur protein had a different relative orientation in different crystal forms, so 2-domain multicrystal averaging was used with different rotation-translation operators for the ironsulfur extrinsic domain and the rest of the protein. Surprisingly the NCS operators for the ironsulfur protein region refined to be the same as for the rest of the monomer, despite asymmetric crystal contacts on the ironsulfur protein. The intercrystal rotation-translation operators for the ironsulfur domain were improved by optimizing operators to take the calculated density from the known structure(11) of the soluble domain of this protein into the density of each crystal's map, maximizing the correlation coefficient. This correlation coefficient also served as an unbiased criterion for map quality, guiding our strategy for phase improvement. After the psuedo-NCS operator for relating subunits 1 and 2 was determined, its correlation coefficient was also used in this way. Phase improvement was restarted from the isomorphous replacement phases using 2-domain averaging with the new operators.
Phase improvement was repeated many times, using the improved phases from the previous cycle to prepare more accurate masks and operators for the next cycle. Once this process had converged at 4.0 Å, phase extension in gradual increments was carried out, increasing the resolution limit by adding 1/(240 Å) to 1/dmin and performing 4 cycles of 2-domain multicrystal + NCS averaging to phase the newly added reflections before making the next resolution increment.
Figure Legends
Figure 1. Improvement and extension of phases by molecular averaging. Electron density at different stages and from different crystals in the region of the cytochrome c1 heme. a: isomorphous phases. b: phases improved by multicrystal averaging. c, d: phases extended to include all data. a-c: crystal used for isomorphous phasing (labeled chk in Table 3a) d: higher-resolution dataset from crystal chc.
Figure 2. Ribbon diagram of the bc1 dimer from our model coordinates.
Figure 3. Transverse sections of the ribbon diagram of Figure 2 at three different levels.
a. In the transmembrane section at the level of the high potential hemes. b. at the level of the high potential hemes. c. In the inter-membrane (external surface) domains. Fig.3d. Secondary structure assignment for cytochrome b.
Figure 4. The regions around the hemes of cytochrome b./ The environments of
the hemes of cytochrome b. The maps are from experimental phases improved
and extended to 3.0 Å by averaging over multiple crystal forms and
non-crystallographic symmetry as described in Table 2. The maps are contoured
at 2.0 (blue) and 12.0 (orange). The ball and stick figures
are from the current unrefined model, with the backbone of cyt. b in red and
side chains of cyt. b and heme in CPK colors. A. High potential heme.
Helix B is visible on the upper left but leaves the slab, toward the viewer, in
the lower left. Residues H98 and R101 extend into the slab toward the heme from
helix B. Helix A comes into the slab in the lower left, due to spiraling of the
helices in the 4-helix bundle, and residue W32 reaches from it toward the
viewer. Helix C is visible on the right, at the edge of the slab toward the
viewer. His-197 comes up toward the viewer from Helix D which is out of the
slab away from the viewer.
B. Low potential heme.
Figure 5. Inhibitor binding sites. Inhibitor crystals were phased initially with improved/extended phases of the native crystal for determining molecular replacement parameters, then rephased by molecular replacement using electron density of the native crystal as a model. These phases were further improved by molecular averaging over non-crystallographic symmetry and over other chicken crystals. Blue maps are electron density from the inhibitor-containing crystal. [Orange maps are difference maps, inhibitor minus native]. A. Antimycin. B. Stigmatellin.
Figure 6. Correlation of experimentally-phased electron density with the
atomic model of the Rieske protein. The maps are calculated as in Figure 4
and contoured at 1 (blue) and 5 (orange). The atomic model
of the ironsulfur protein (CPK colors) is from the coordinates of the protein
database entry 1RIE, after rotation and translation of the whole structure to
best fit the experimental density as described in the text. The background of
cytochrome b (red) is from our model. A. A slab through the whole
protein, including ironsulfur cluster and N-terminal residue A70, showing the
density continuing into a transmembrane helix. B. A close-up of the
cluster area.
Figure 7. Attachment of the Rieske Protein (in the distal conformation) to
cytochrome b. The map is calculated as in Figure 4 and contoured at 1
. The atomic model of the ironsulfur protein is from the coordinates
of the protein database entry 1RIE. The model of cytochrome b (red backbone
plus ball-and-stick models for residues W142 and L263 through P266) is from our
coordinates.
Figure 8. Relative positions of the redox centers in the two different conformations. Left- From a native crystal, with Rieske in the distal position. Right- From a crystal with stigmatellin present, with the Rieske protein in the proximal position.
The distances on the left are measured between peaks in the Bivoet difference map at 7131 eV. The distances in the presence of stigmatellin were measured between peaks in electron density maps. Below- both positions are illustrated in a single stereo figure.
Figure 9. Two conformations of the Rieske protein. Ribbon diagrams of the Rieske ironsulfur protein from our native crystal (yellow) superimposed on the structure from crystals grown in the presence of stigmatellin(blue). A ribbon diagram of cytochrome b is shown in blue-green. The iron-sulfur clusters and hemes are shown as ball-and-stick models.
Figure 10. Comparison of the structures of cytochrome c1 and cytochrome c
(stereo pair). Above is a ribbon diagram of mitochondrial cytochrome c in a
standard orientation with the open C corner of the heme facing the viewer, and
the heme propionates directed downward. Below is our preliminary structure of
cytochrome c1, rotated to put the common features between the two cytochromes
in the same orientation. Corresponding segments of each cytochrome are drawn
with the same color. Helices labeled 1, 3, and 5
correspond to similarly labeled helices in cytochrome c, while those labeled
2* and 6* have no counterpart in cytochrome c.
Figure 11. Interface between cytochrome c1 and the Rieske protein. The electron density map is from merged data of two beef hexagonal crystals, phased and improved as described in Table 2. The ironsulfur model (left) is from the protein database file 1RIE, positioned as described in the text. The cytochrome c1 model (right) is from our chicken bc1 model, positioned with the same operator used to position the main part of the bc1 complex in these crystals.
Figure 12. Electron pathway through cytochrome c1 in a hypothetical complex
of ubiquinol:cytochrome c oxidoreductase with cytochrome c. C
traces represent the backbones of cytochrome c1 (green), cytochrome c (red) and
the Rieske protein (red). The hemes and the ironsulfur cluster and surrounding
residues are drawn as ball-and-stick models. The distances indicated between
cofactors are between closest atoms in the -bonded system from the native
chicken crystal structure. The position and orientation of cytochrome c are
from preliminary analysis of co-crystals which have cytochrome c bound to one
monomer, however this position has not yet been established reliably.
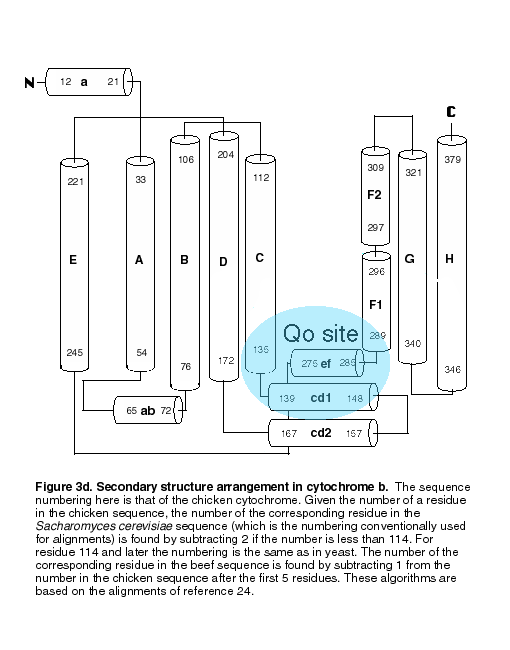{kind=link}
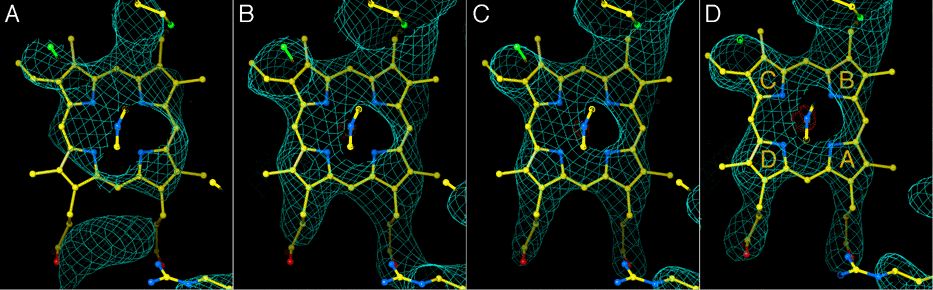{kind=link}
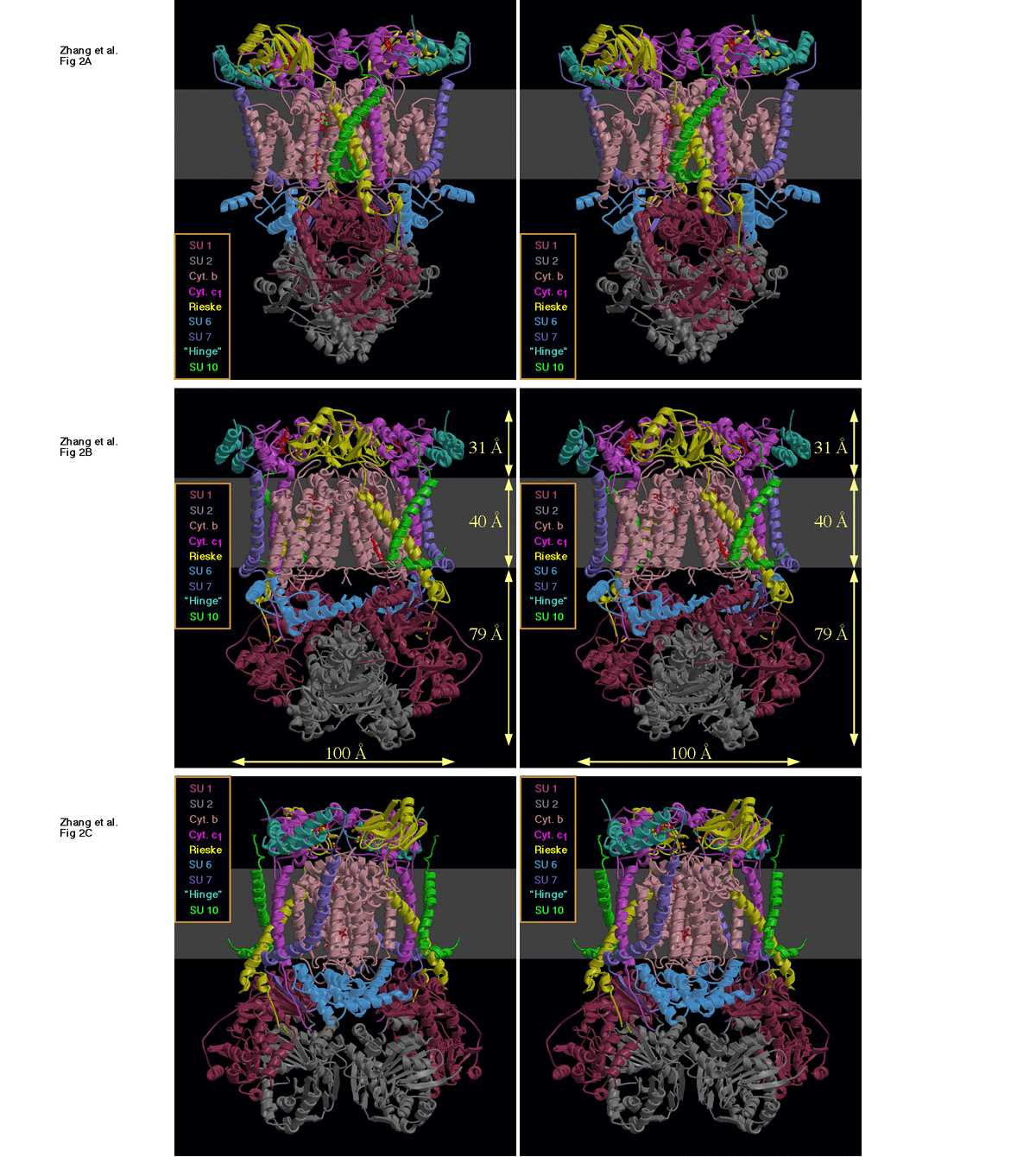{kind=link}
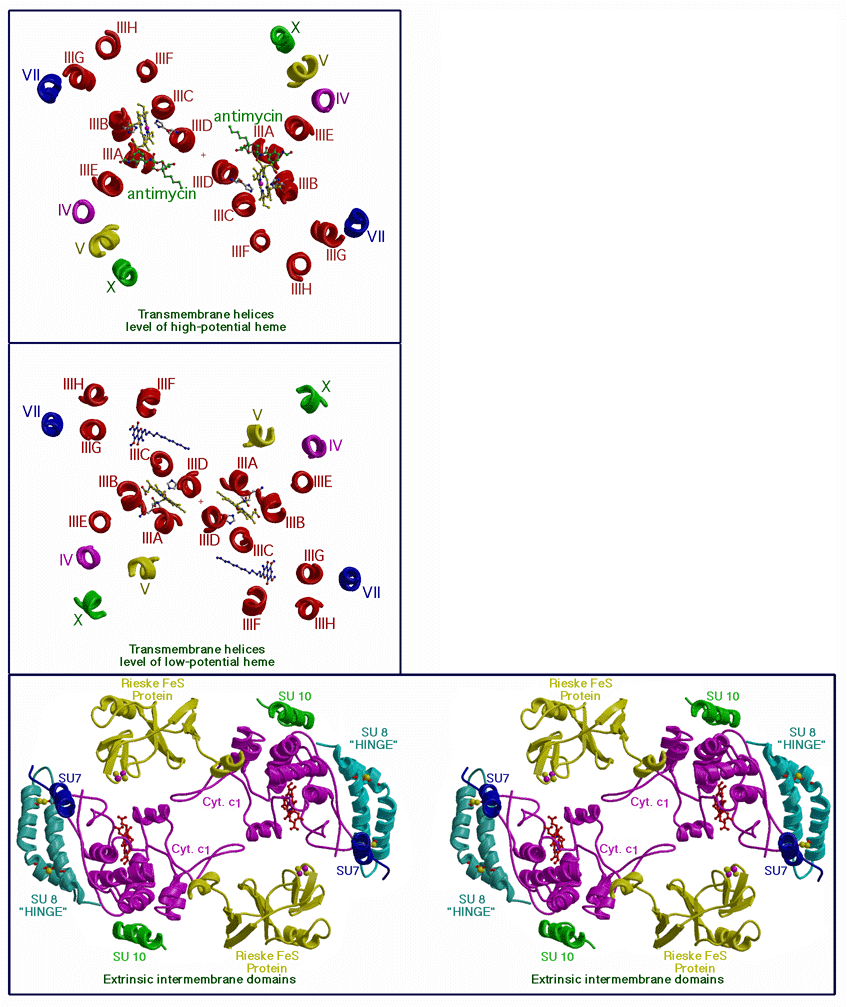{kind=link}
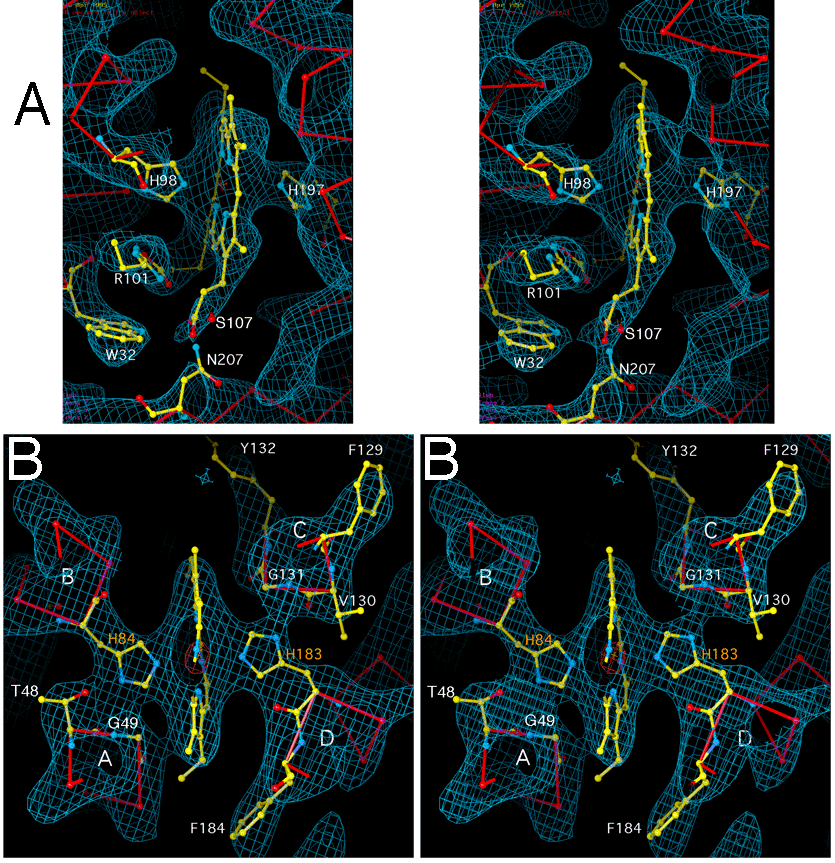{kind=link}
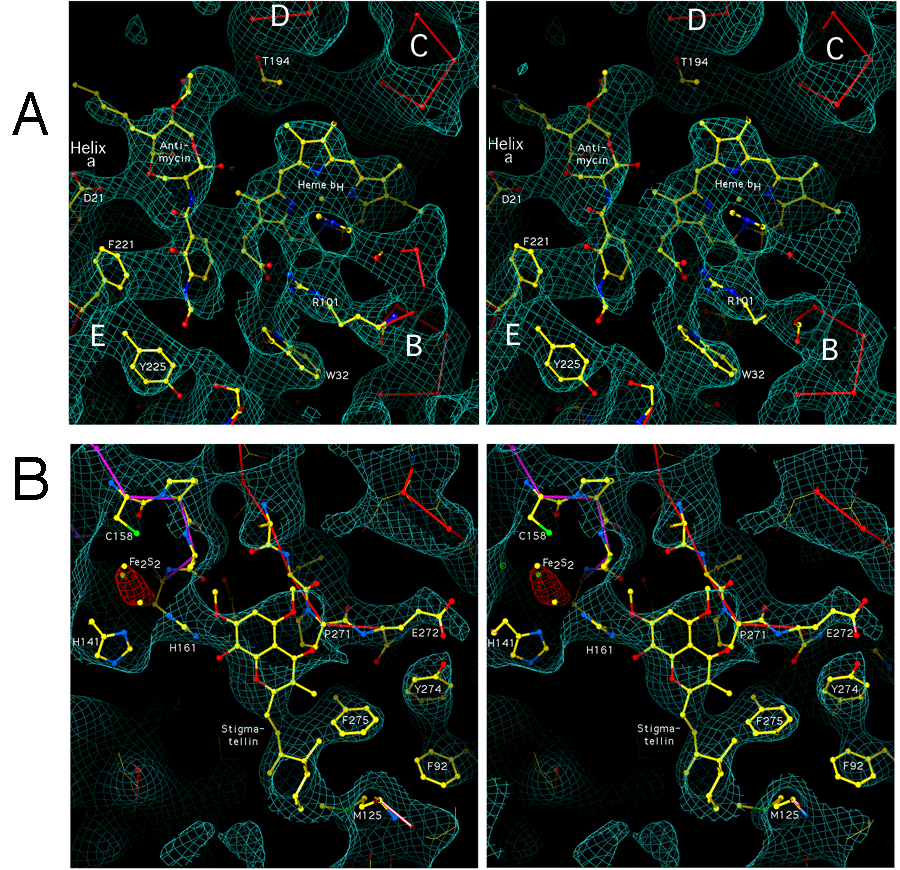{kind=link}
{kind=link}
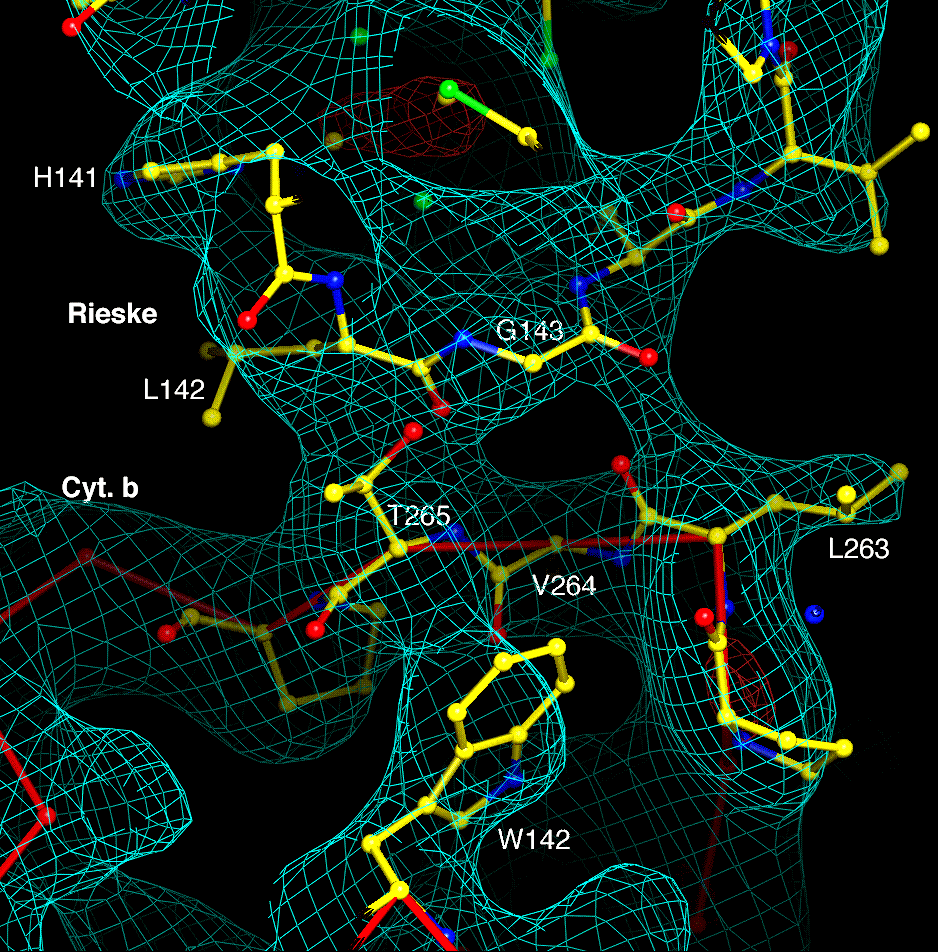{kind=link}
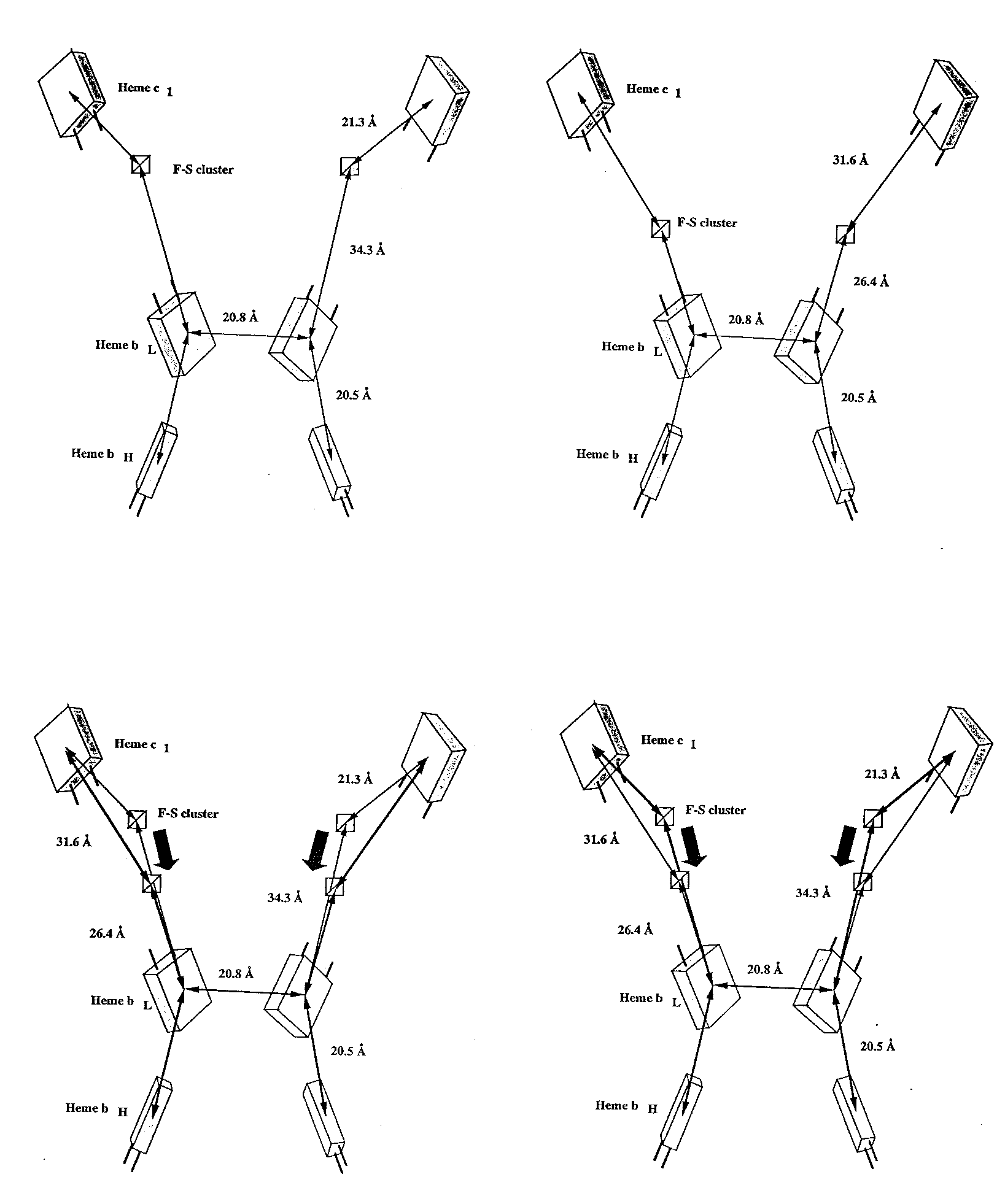{kind=link}
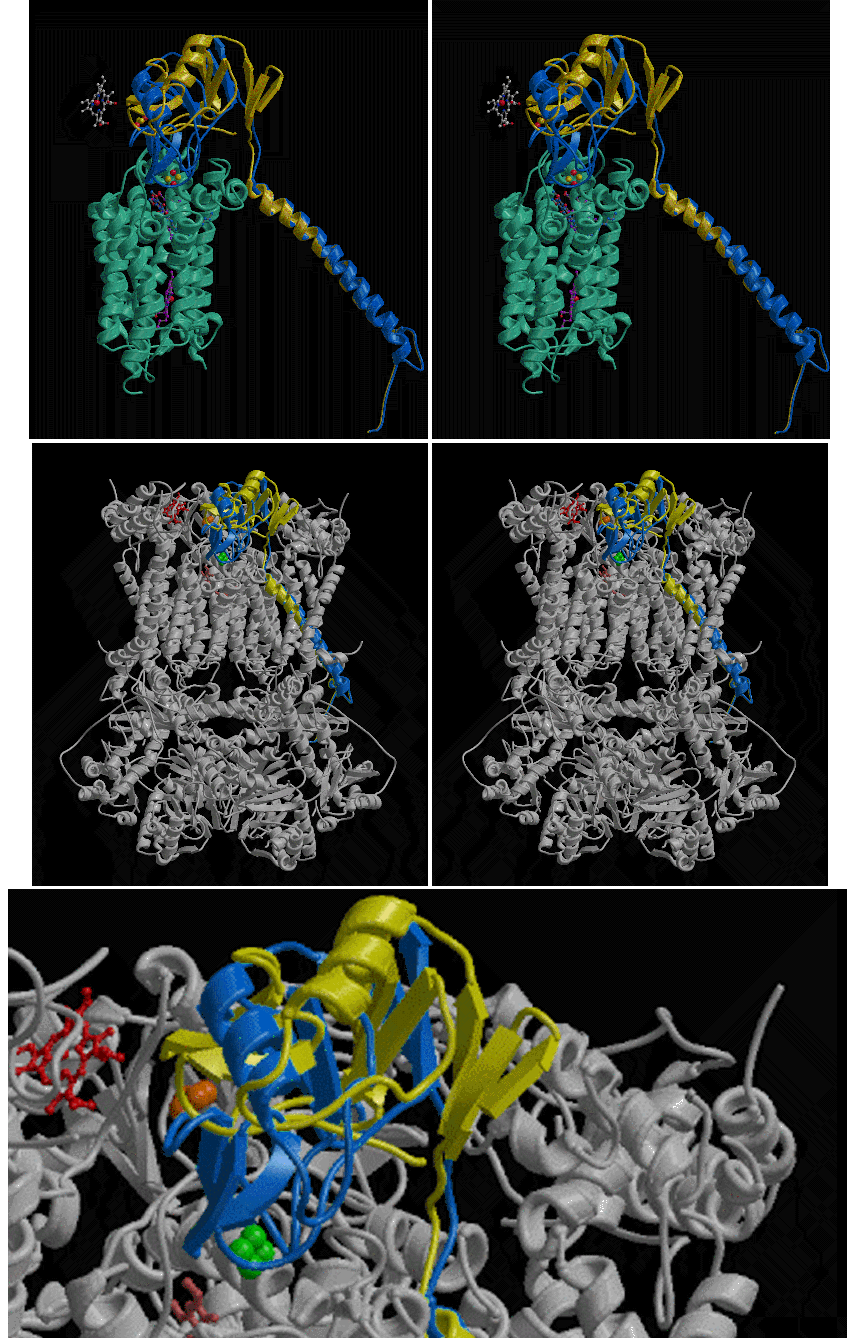{kind=link}
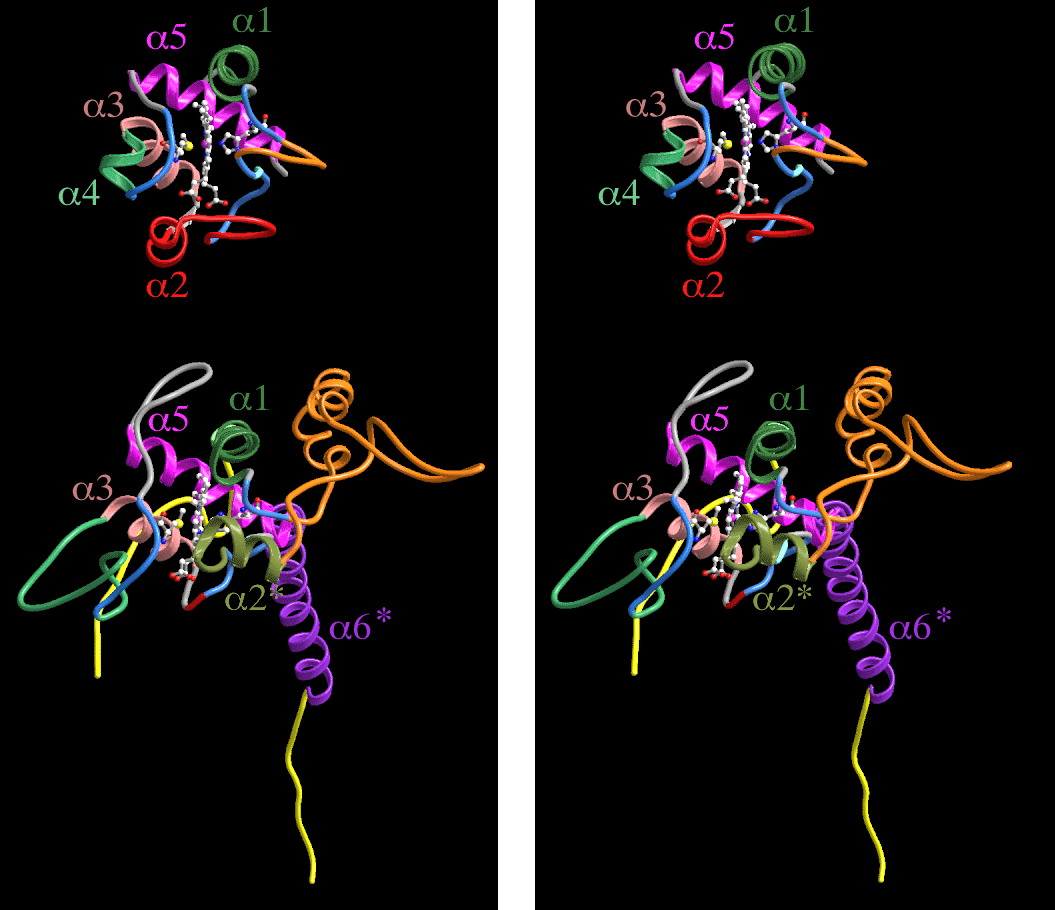{kind=link}
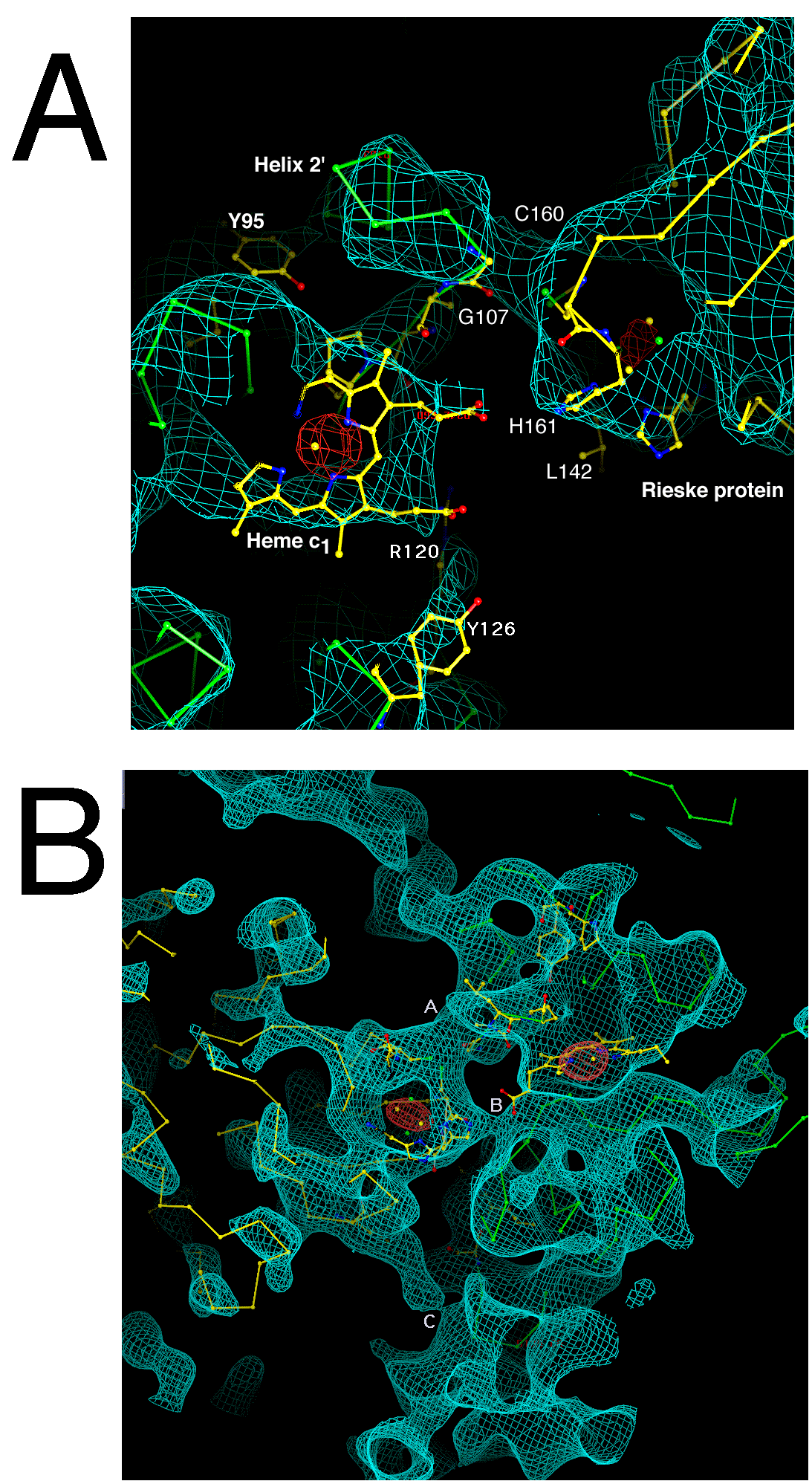{kind=link}
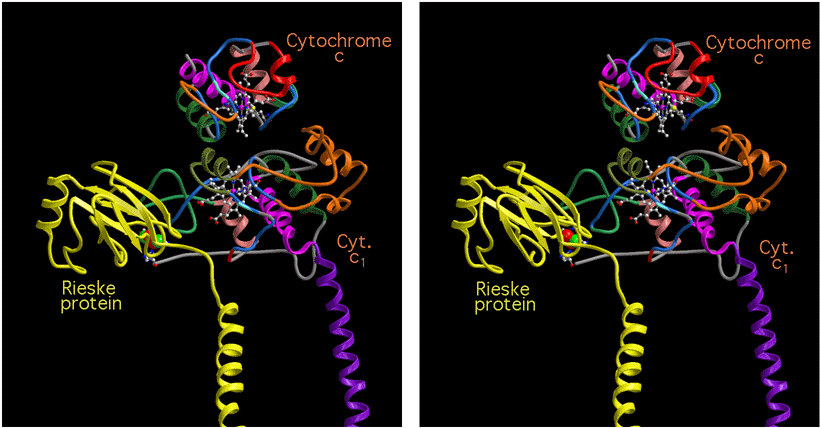{kind=link}